ニューラルネットワーク分子動力学法(NNMD)は第一原理計算よりも高速で既存の分子動力学計算よりも高精度なシミュレーションが可能となりますが、 NNMDで使用する原子間ポテンシャルの作成には第一原理計算を用いた大量の教師データが必要となることが課題となっています。 本事例では、クラウドシステムを利用して一度に大量の第一原理計算を同時実行して学習データを作成することで、短時間でポテンシャル学習を行い、 従来力場では再現が困難なCeO2の固液界面の挙動を、NNMDを用いて解析した事例を紹介します。
1. NNMDポテンシャルのための学習データ作成
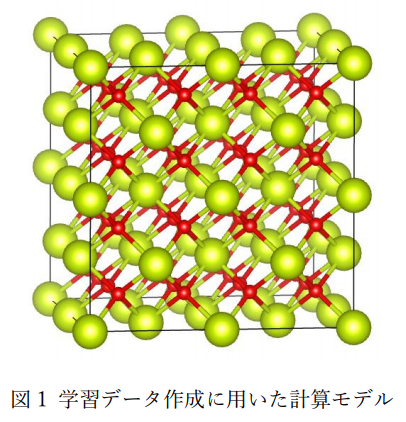
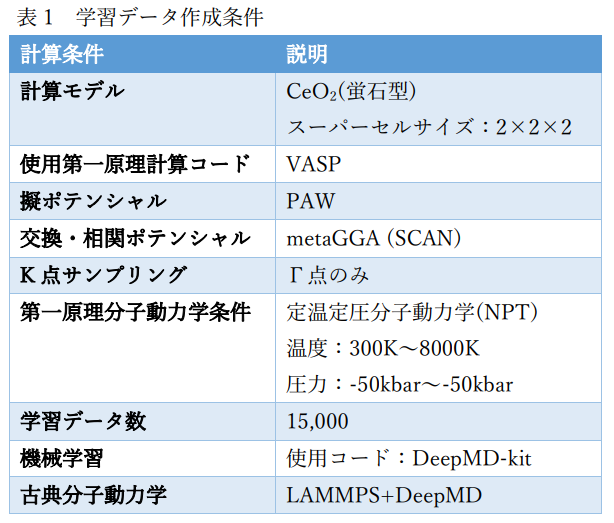
図1にCeO2の学習データ作成に用いた計算モデルを示します。また、表1にその計算条件の詳細を示します。 ポテンシャルの機械学習にはDeepMD-kit[1]を使用しました。
2. 計算結果
①比熱および熱膨張係数

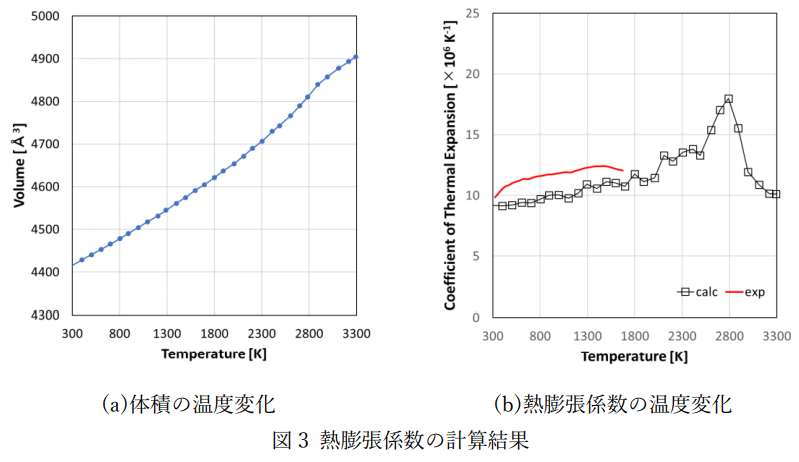
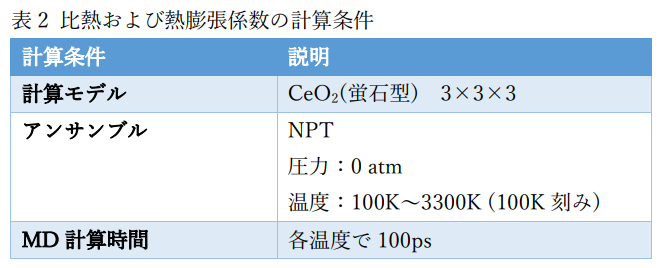
作成したポテンシャル関数および分子動力学コードLAMMPSを用いて、CeO2の比熱および熱膨張係数を計算しました。 計算条件を表2に示します。比熱および熱膨張係数はエントロピーおよび体積の温度微分を用いて評価しました。 エントロピーの温度変化および比熱の計算結果を図2に示します。また、体積の温度変化および熱膨張係数の計算結果を図3に示します。 計算結果は実測[2]とよく対応していることが分かります。
②融点


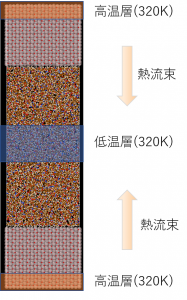
図2および図3の比熱および熱膨張係数の計算では、融点を正確に評価できていないため、固液共存モデルを用いて融点の評価を行いました。 融点の計算条件および計算モデルを表3および図4に示します。
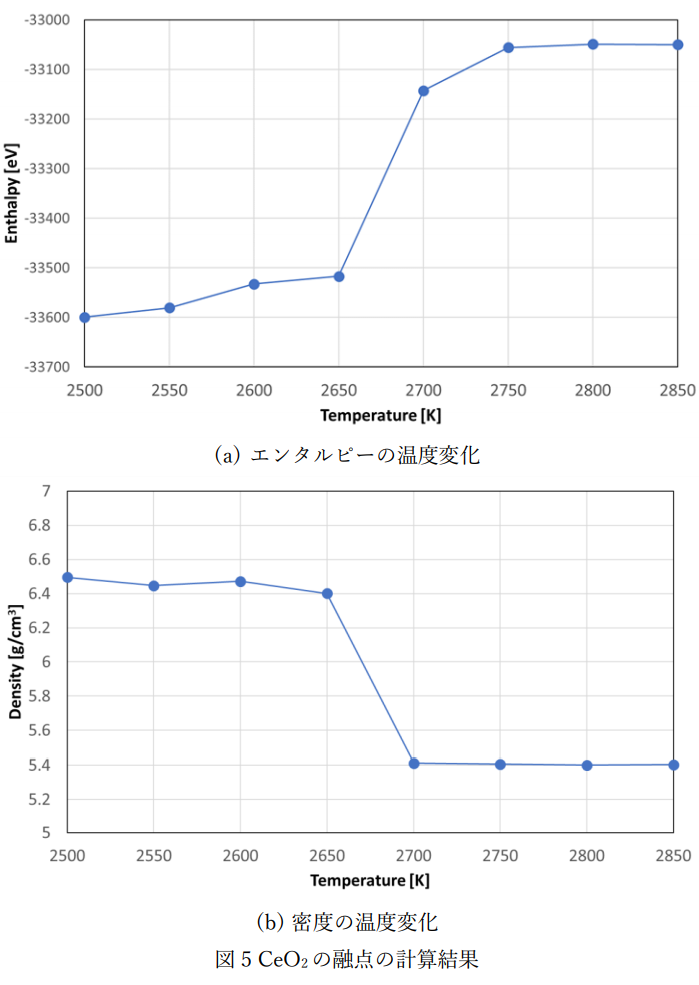
図5にエンタルピーおよび密度の温度依存性の計算結果を示します。エンタルピーおよび密度はCeO2の実測の融点の近傍2650K~2750Kで不連続となり、 融点付近で固相と液相が共存していることが確認できました。
融点近傍の2650Kと2700KにおけるMDアニメーションを左に示します。2650Kでは液相が結晶化しているのに対して、2700Kでは固相が溶融している様子が確認できています。
3.参考文献
[1]Han Wang, Linfeng Zhang, Jiequn Han, and Weinan E. “DeePMD-kit: A deep learning package for many-body potential energy representation and molecular dynamics.” Computer Physics Communications 228 (2018): 178-184.
[2] N. Nelson, D Rittman, J. White et al., “An Evaluation of the Thermophysical Properties of Stoichiometric CeO2 in Comparison to UO2 and PuO2”, Journal of the American Ceramic Society, 97, 3652-3659 (2014)