ニューラルネットワーク分子動力学法(NNMD)は第一原理計算よりも高速で既存の分子動力学計算よりも高精度なシミュレーションが可能となりますが、 NNMDで使用する原子間ポテンシャルの作成には第一原理計算を用いた大量の教師データが必要となることが課題となっています。 本事例では、クラウドシステムを利用して一度に大量の第一原理計算を同時実行して学習データを作成することで、短時間でポテンシャル学習を行い、 従来力場では再現が困難なCeO2の固液界面の挙動を、NNMDを用いて解析した事例を紹介します。
1. NNMDポテンシャルのための学習データ作成
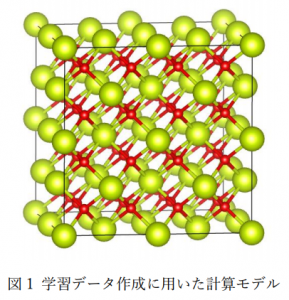
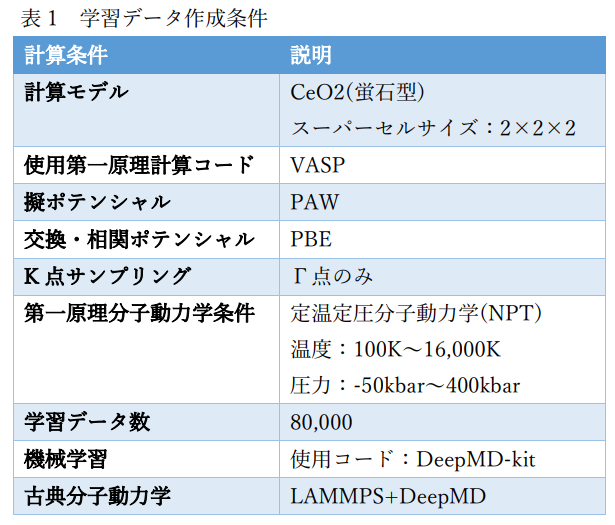
図1にCeO2の学習データ作成に用いた計算モデルを示します。また、表1にその計算条件の詳細を示します。 ポテンシャルの機械学習にはDeepMD-kit[1]を使用しました。
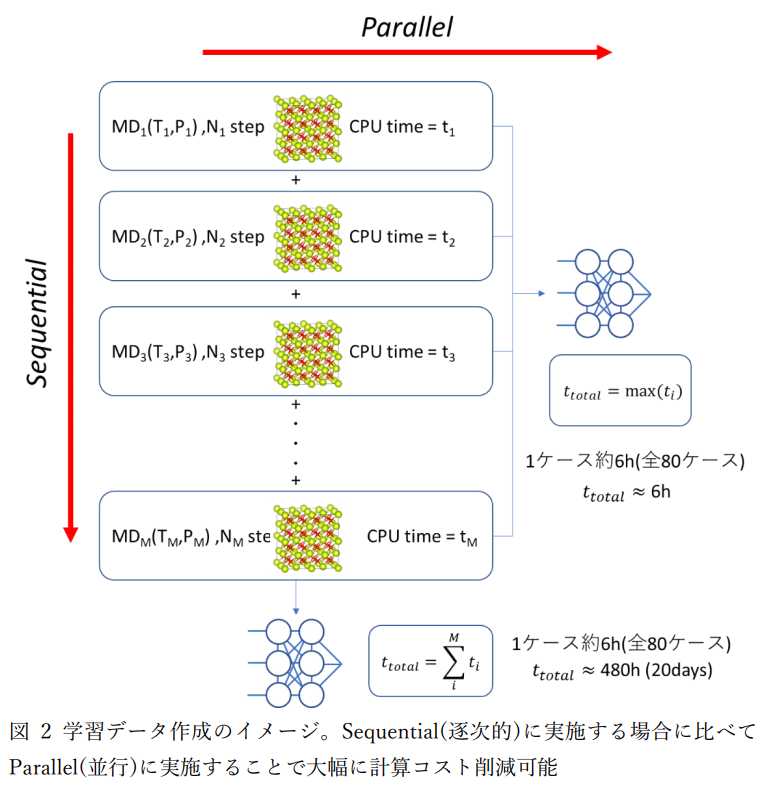
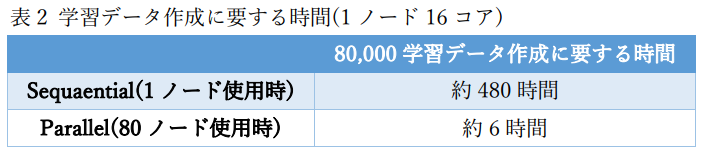
本事例では学習データの作成のために、広範囲の温度・圧力条件を課した第一原理分子動力学計算を多数回実行したため、 通常のクラスター計算機環境で逐次的に計算を実行すると膨大な計算時間が必要となりますが、 クラウドシステムを使用することで、一度に全ての第一原理計算度同時に実行して短時間でポテンシャル学習を行うことができました(図2,表2)。 今回、80,000個の学習データの計算に第一原理分子動力学計算を80ケース実行しています。
2. ベンチマーク計算

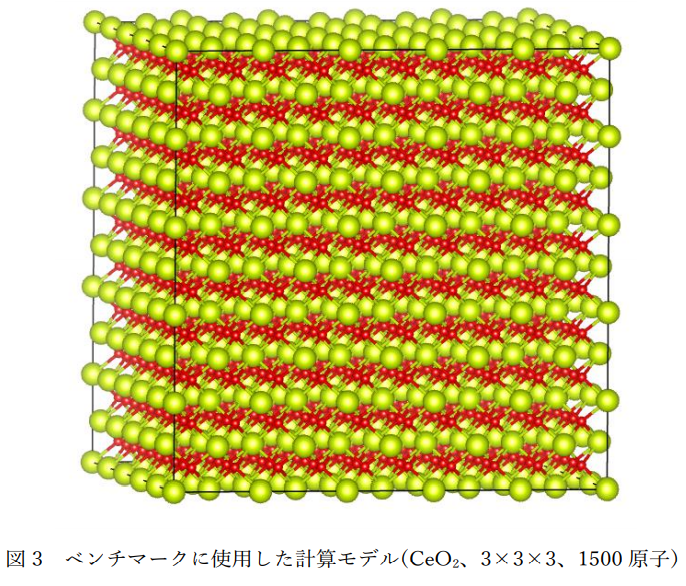

本事例で使用したDeepMD-kitはGPUに対応しており高速にポテンシャルの学習を行うことが可能です。 また、作成したポテンシャルパラメータは無償の分子動力学計算コードLAMMPSで使用可能となっており、 MD計算もMPI並列およびGPU高速化に対応しています。表3に表1の80,000個の学習データを学習させる場合の計算時間をCPUとGPUで比較した結果を示します。 ポテンシャル学習において、GPUはCPUの約3~4倍の高速化が可能であることが分かります。
また図3のモデルを用いて、MD計算についても同様にCPUとGPUで速度比較した結果を表4に示します。 MD計算においては、GPUはCPUの約10倍高速化されていることが確認できました。
3. CeO2の固液界面のシミュレーション
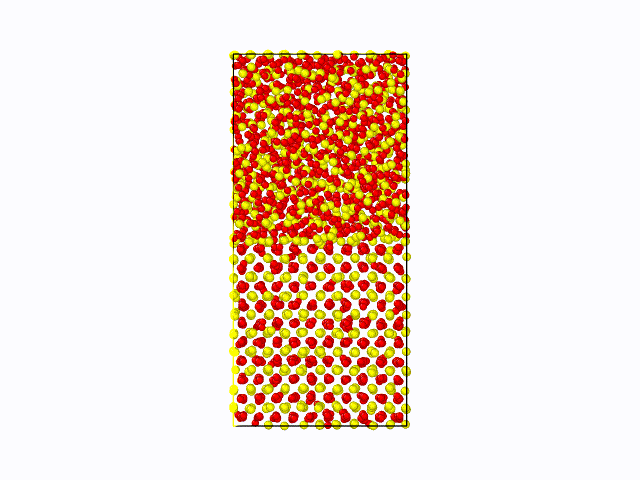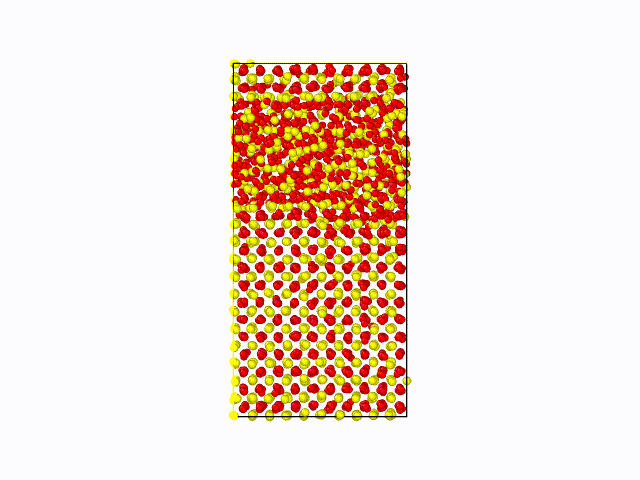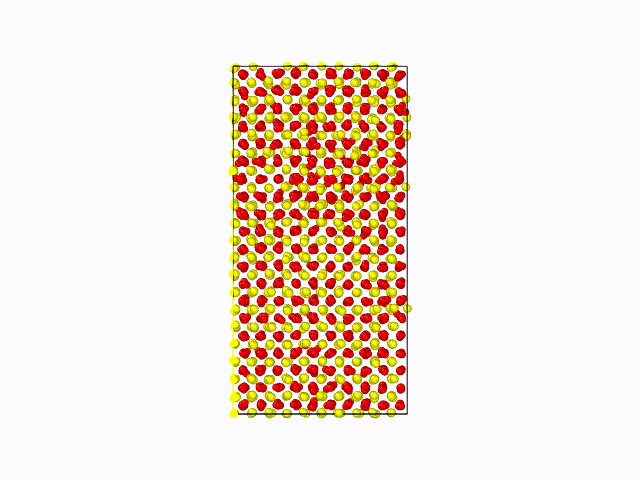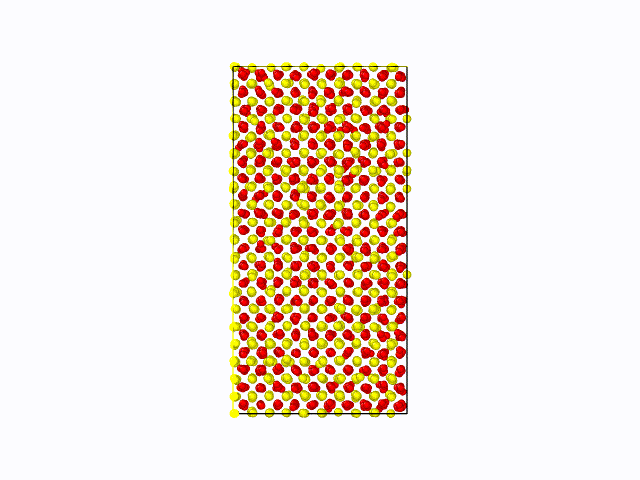
作成したポテンシャル関数を用いて、CeO2の固液界面のシミュレーションを実施した結果を以下に示します。 固相/液相でそれぞれ1500原子(総原子数3000原子)の計算モデルを用いて、1atm、2000Kにて500psのMD計算を実施しました。 液相が界面から徐々に固相へと変化している様子が確認できました。これにより、第一原理計算では計算コストが大きく、かつ力場パラメータが存在せず実施困難な現象も、 NNMDを利用することで計算可能となることが期待できます。
4.参考文献
[1]Han Wang, Linfeng Zhang, Jiequn Han, and Weinan E. “DeePMD-kit: A deep learning package for many-body potential energy representation and molecular dynamics.” Computer Physics Communications 228 (2018): 178-184.