概要
K. Epharaim Babu等がフルポテンシャルでWIEN2Kを用いて、ぺロブスカイト(perovskite)構造のCsCrF3を計算[1]しています。そこで、擬ポテンシャルでの計算で、Exabyte.io とQuantum ESPRESSOを用いて再現できるか検証を行いました。
1. モデル作成
モデル作成を行なうにあたり、結晶構造はMaterials Projectから結晶構造をインポートして作成します。Exabyte.ioの項目のMaterialsで、、「CsSrF3」と入力するだけで、Materials Projectのデータベースから検索することができます。
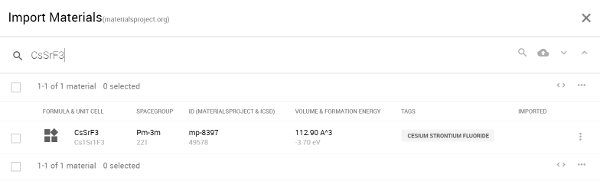
図1 Materials Projectから「CsSrF3」を検索
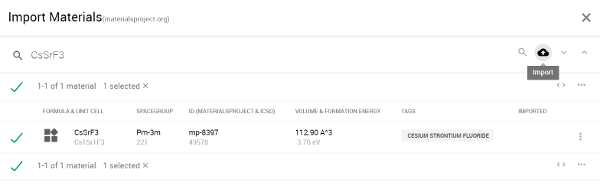
図2 Materials Projectから「CsSrF3」をインポート
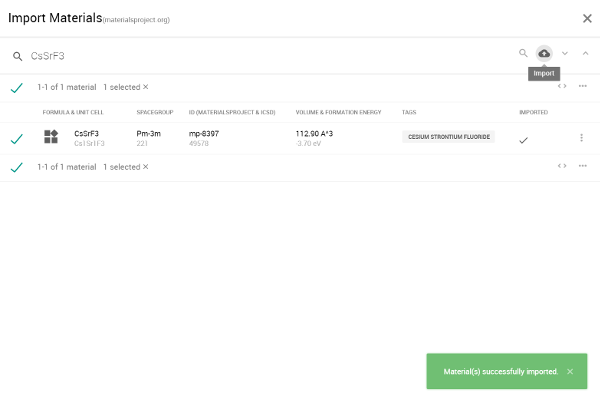
図3 インポート成功の様子
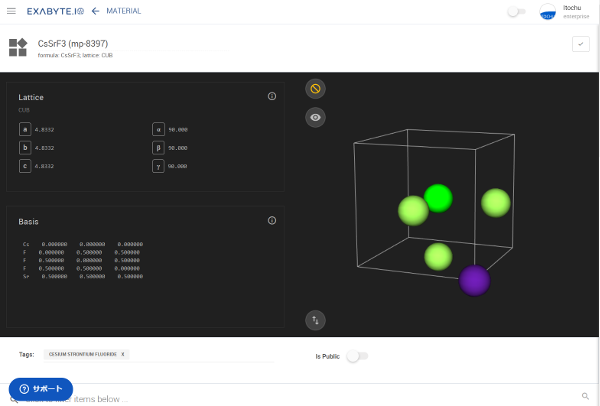
図4 CsSrF3を可視化
2. 計算
2.1. 格子定数最適化
CsSrF3の格子定数を最適化には、ワークフロの「Variable-call Relaxation」を用います。使用する擬ポテンシャルは、Exabyte.ioでデフォルト設定になっているものを用います。
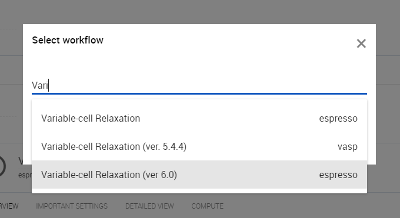
図5 Variable-call Relaxationを検索および選択
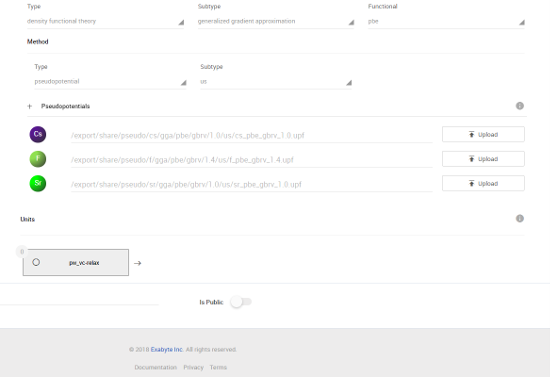
図6 用いる擬ポテンシャルリスト
表 1 格子定数の比較
| 格子定数 [Å] | |
|---|---|
| 実験値 | 4.83 |
| WIEN2K [1] | 4.83 |
| Exabyte.io & Quantum ESPRESSO | 4.82 |
計算結果を比較します。擬ポテンシャルを用いた場合でも実験値やフルポテンシャルの結果と概ね一致する結果でした。
2.2. バンドギャップ
図7 ワークフロからBand Gapを選択
バンドギャップを求める際には、ワークフロの「Band Gap」を用います。
計算の際に、「parent」機能を使うと、格子定数を最適化したジョブの結果を元にバンドギャップの計算を行うことができます。
図8 「parent」機能
表 2 バンドギャップの比較
| Direct band gap [eV] | |
|---|---|
| WIEN2K [1] | 6.34 |
| Exabyte.io & Quantum ESPRESSO | 6.26 |
計算結果を比較します。フルポテンシャルの結果に近い結果でした(表2)。
2.3. バンド分散
バンド分散を求める際には、ワークフロの「Band Structure」を用います。k点パスも自動設定されます。
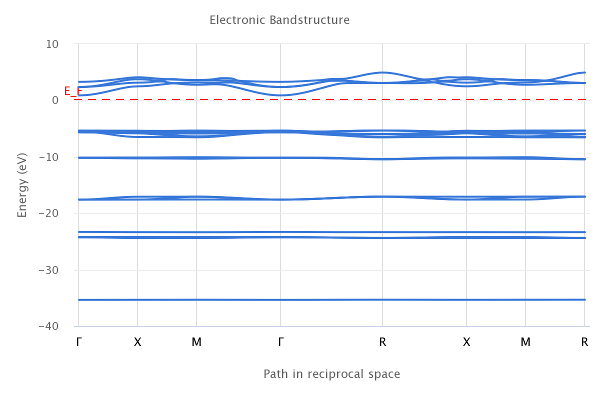
図9 バンド分散の結果
2.4. 状態密度
状態密度(DOS)を求める際には、ワークフロの「Density of States」を用います。トータルの密度以外に、各軌道でおこなうPDOSの計算も同時に行います。

図10 状態密度の計算
3. 参考文献
[1] K. EPHRAIM BABU, A. VEERAIAH, D. TIRUPATHI SWAMY, V. VEERAIAH, “First-principles study of electronic and optical properties of cubic perovskite CsSrF3”, Materials Science-Poland, 30 (2012), 359-367