通常の分子動力学法ではナノ秒オーダーの解析が限界ですが、それよりも長い時間スケール の問題への対策として、時間スケールを加速させる種々の加速分子動力学法[1]が提案されています。 本手法は、半導体・誘電体、電池、鉄鋼材料中の欠陥/不純物拡散や転移生成、表面/界面における元素 拡散や結晶成長など、種々の問題への応用が期待できます。ここでは長時間シミュレーションが必要な事例 としてα鉄中の炭素拡散の検証 を行いました。α鉄中の炭素拡散は隣接する八面体中心のサイト間の移動 に室温で0.1秒程度かかるため通常のMD法では拡散を再現できませんが、加速分子動力学法を用いること で長時間のシミュレーションが可能となり、α鉄中の炭素拡散の様子を確認することができました。
1. 計算モデルおよび計算条件

図1 使用するモデル
表1 計算条件
左に計算に使用したモデルおよび計算条件を示します。加速分子動力学法として、 今回は温度加速分子動力学(temperature accelerated dynamics :TAD)を用いました[2]。 TAD法では短時間の高温のシミュレーションから原子配置のサンプリングを行い、各原子配置間の遷移確率を計算することにより シミュレーションを加速させる方法です。本シミュレーションでは鉄の融点の2倍程度の温度を高温条件に設定しました。 全ての計算は分子動力学計算コードLAMMPSを用いて実施しました。
2. 計算結果
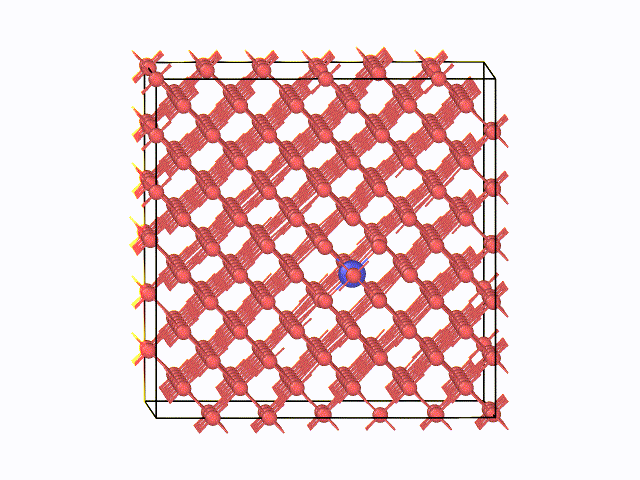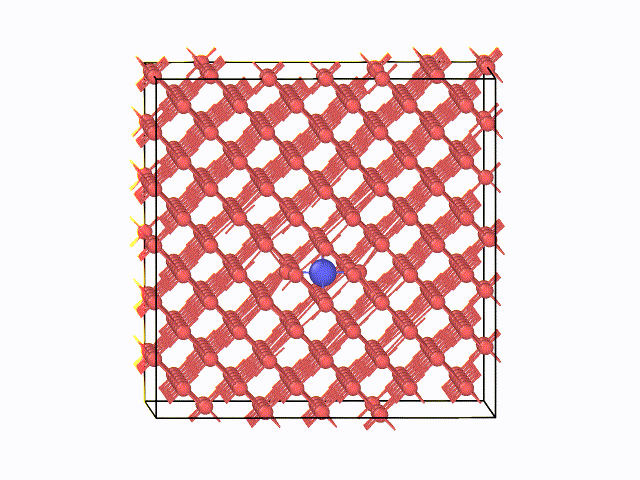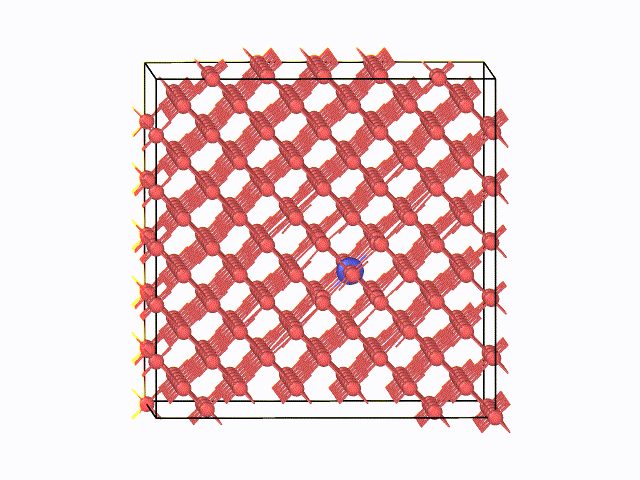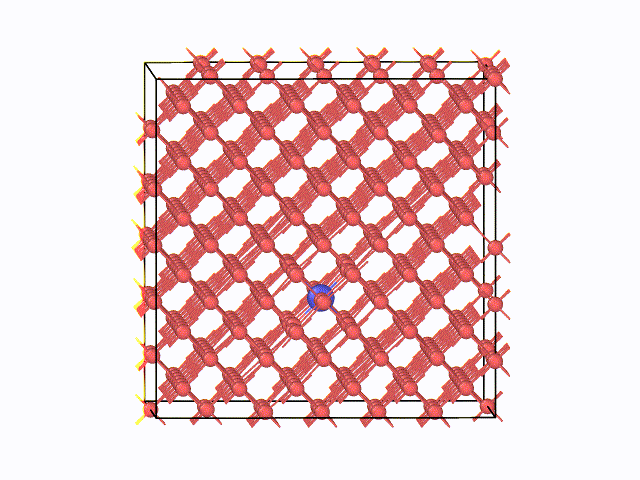
図2 423K(150℃)におけるMDスナップショット
左に、423K(150℃)におけるMDスナップショットを示します。α鉄の八面体中心間を炭素原子がジャンプする様子が 確認できています。
表2 温度と平均ジャンプ時間
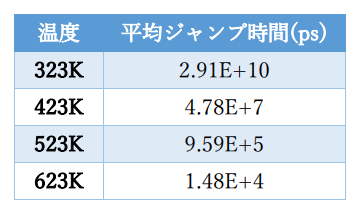
各シミュレーション温度における平均ジャンプ時間を示します。初期条件を変えた5回のシミュレーション結果を用いて、 炭素原子がα鉄の八面体中心をジャンプする時間の平均値を評価して平均ジャンプ時間としました。 平均ジャンプ時間は低温では数μ秒程度となるため、通常のMD法では再現できない時間スケールのシミュレーションが実施できている ことが分かります。
さらに、炭素原子の平均二乗変位および拡散係数の計算結果を示します。拡散係数の計算結果は実測値[4]を再現できていることが 分かります。通常のMD計算ではこのような低温時の元素拡散をシミュレーションすることは困難なため、本手法が非常に有用 となります。
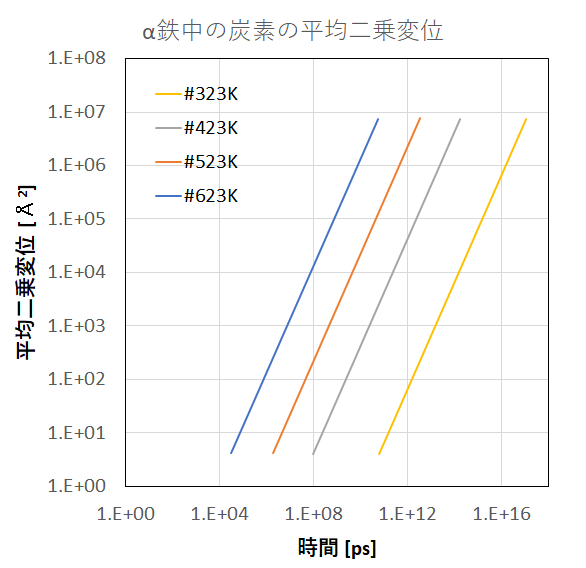
図3 α鉄中の炭素の平均二乗変位
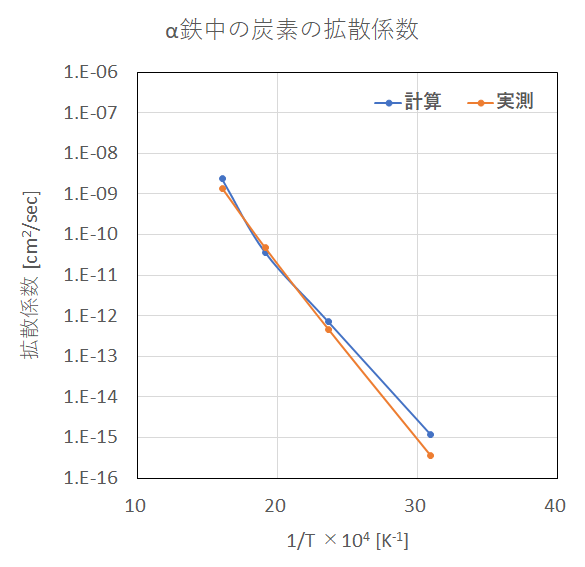
図4 α鉄中の炭素の拡散係数
3. 本手法の適用分野
今回は鉄鋼分野における加速分子動力学法の適用事例を紹介しましたが、その他、転移生成や欠陥移動など長時間シミュレーションが 必要となる現象、分野への応用が期待できます。以下に、本手法の適用が可能性な分野の一例を紹介します。
①半導体・誘電材料中の不純物・欠陥の拡散
②電池材料中のイオン拡散(従来のMD計算では評価困難な実動作温度でのイオン拡散の評価が可能)
③表面/界面における元素拡散、結晶成長
4.参考文献
[1]Arthur F Voter, Francesco Montalenti, Timothy C Germann. “Extending the time scale in atomistic simulation of materials” Annual review of materials research, 32, 321-346, (2002)
[2] Mads R Sorensen, Arthur F Voter, “Temperature-accelerated dynamics for simulation of infrequent events”, The Journal of Chemical Physics, 112, 9599-9606 (2000)
[3] 邦武 立朗, “鉄および鋼中の炭素の拡散”, 日本金属学会会報, 3巻9号, 466-476 (1964)
[4] Kim H-K, Jung W-S, Lee B-J, “Modified embedded-atom method interatomic potentials for the Fe–Ti–C and Fe–Ti–N ternary systems”, Acta Materialia, 57, 3140 (2009)