ここでは孤立分子の吸収スペクトルを時間依存密度汎関数法(TDDFT)で計算する方法を説明します。例としてベンゼン分子を用います。
1.計算モデル作成
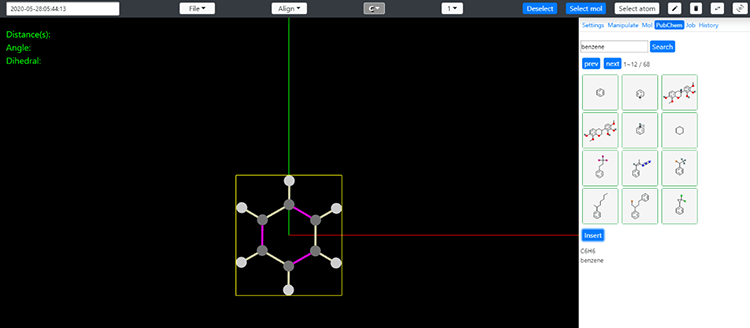
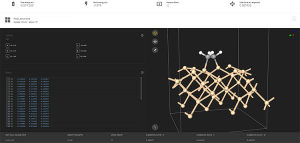
図1 MI-nanosysを用いた分子モデリング
分子のモデリングは弊社で作成したMI-nanosysを併用することで作成可能です。MI-nanosysはExabyte.ioのアカウントで使え、さらに無料で使用することができます(図1)。

図2 MI-nanosysよりExabyte.ioへモデルの転送
MI-nanosysから作成したモデルをExabyte.ioのMATERIALSに転送します。
2.TDDFTの実行
表1 TDDFTの計算条件
| 項目 | 詳細 |
|---|---|
| 擬ポテンシャル | C_ONCV_PBE_sr.upf H_ONCV_PBE_sr.upf |
| カットオフ 波動関数 | 40 Ry |
| カットオフ 電子密度 | 160 Ry |
| k点 | scf:4×4×4 |
| 収束閾値 | 10-10 |
TDDFTを用いた孤立分子の吸収スペクトルの計算はturbo_lanczos.xおよびturbo_spectrum.xを用いて実行します。TDDFTの計算条件は以下の通りです。

図3 吸収スペクトルの計算結果
図3にTDDFTを用いたベンゼン分子の吸収スペクトルの計算結果を示します。計算結果は実験結果[1]と非常によく一致していることが確認できています。
3.TDDFTのワークフローについて
Quantum EspressoのTDDFTで吸収スペクトルを計算するためのワークフロー(図4)と詳細情報(表2)を示します。
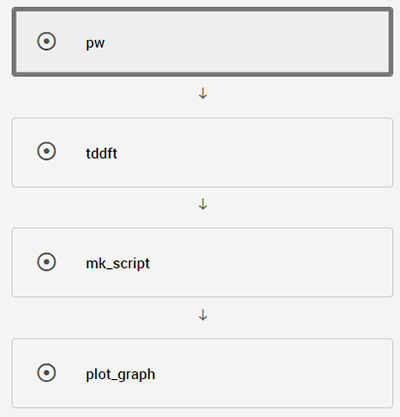
図4 TDDFT計算のワークフロー
表2 TDDFTのワークフロー情報
| 項目 | 説明 |
|---|---|
| ワークフロー名 | absorption_spectrum_for_molecule(QE6.3) |
| 制限事項 | 孤立系のみ |
| フロー内容 pw tddft mk_script plot_graph | SCF計算のフロー TDDFT計算のフロー グラフ描画のスクリプト作成フロー グラフ描画のスクリプト実行フロー |
| 出力ファイル spectrum.csv spectrum.png | グラフデータのCSV出力 吸収スペクトルのグラフ |
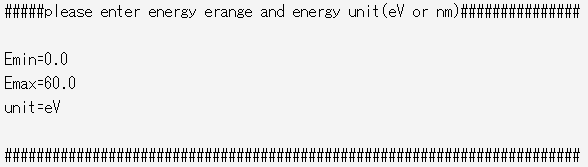
図5 グラフ描画実行フローplot_graph中のエネルギー範囲のおよび単位の入力。Eminがエネルギー下限値、Emaxがエネルギー上限値、unitが使用単位(eV or nm)に対応
また、グラフ出力のエネルギー範囲およびエネルギー単位はplot_graphフロー中に入力することで設定可能です(図5)。
TDDFTのワークフローはBankから「absorption_spectrum_for_molecule(QE6.3)」を取得してご利用ください。
4.計算時間とコスト
最後に本計算の実施にかかった計算時間とコストを表3に示します。
表3 TDDFTを用いた吸収スペクトルの計算時間とコスト(セービングノードを使用)
| 分子 | 計算時間 | 金額 | コア数 |
|---|---|---|---|
| C6H6 | 約7分 | $ 0.1 | 36 |
参考文献
[1] Renfei Feng, Glyn Cooper and C.E Brion, "Dipole (e,e) spectroscopic studies of benzene: quantitative photoabsorption in the UV, VUV and soft X-ray regions", J. Electron. Spectros. Relat. Phenomena, 123, 199-209 (2002)