汎用型グラフニューラルネットワーク力場(GNN力場)を用いて触媒材料の分子動力学シミュレーションを行いました。用いたGNN力場は、Facebook社とカーネギーメロン大学が主催するOpen Catalyst Project[1]で開発されたもので、触媒材料をターゲットとして、約1億3千万個の第一原理計算の計算結果を学習データとして構築されており、広範囲な触媒材料の分子動力学計算が可能となっています。本事例では、白金触媒表面上へのCO分子の吸着ポテンシャルカーブを、GNN力場を用いて計算し、第一原理計算結果と比較することでGNN力場の精度の確認を行いました。
計算モデル・計算条件
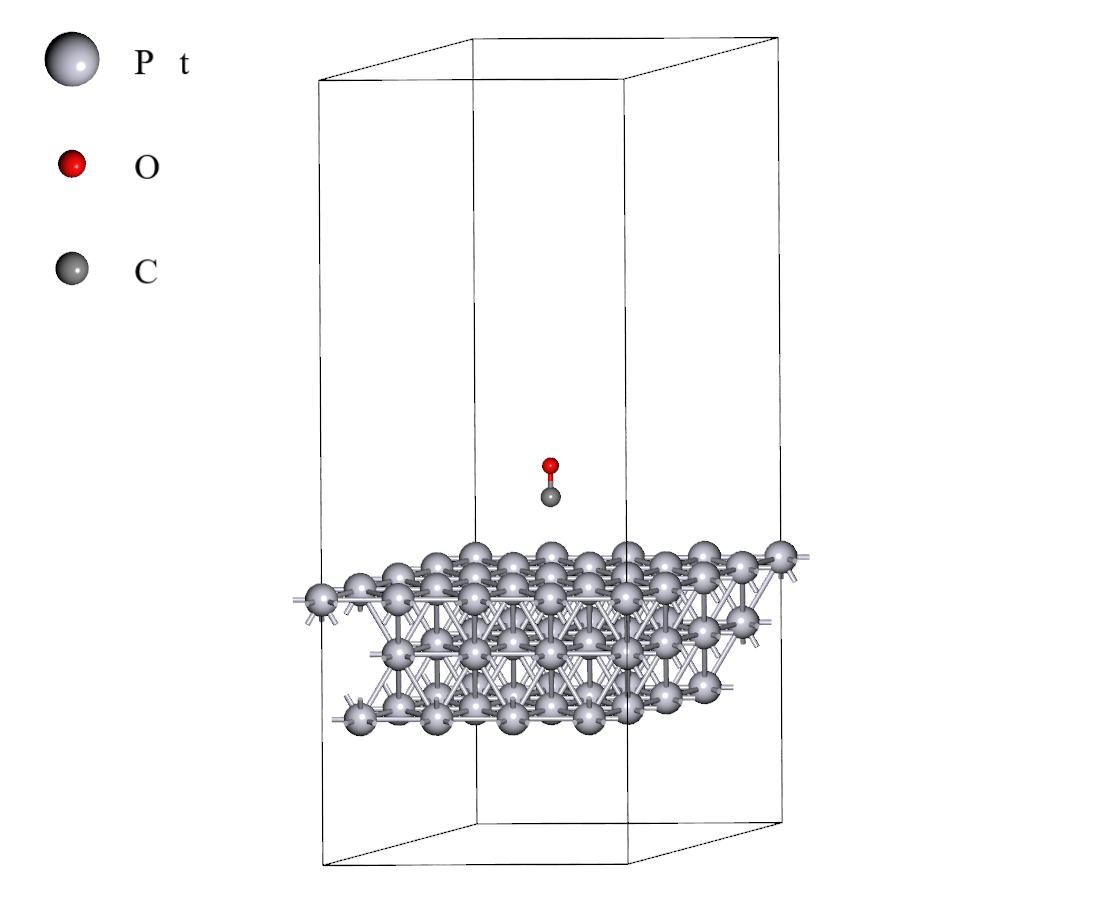
白金触媒表面上へのCO分子吸着は図1に示す計算モデルを用いて計算しました。また、表1に第一原理計算条件および使用したGNN力場の手法を示します。
| 第一原理計算条件 | GNN計算条件 | ||
| 使用コード | VASP (ver 5.4.4) | 使用コード | LAMMPS[2] |
| 交換相関汎関数 | RPBE | 使用GNN | Gemnet+dT |
| 擬ポテンシャル | PAW | Dimnet++ | |
| カットオフエネルギー | 400eV | Schnet | |
| k点サンプリング | 3×3×1 |
計算結果
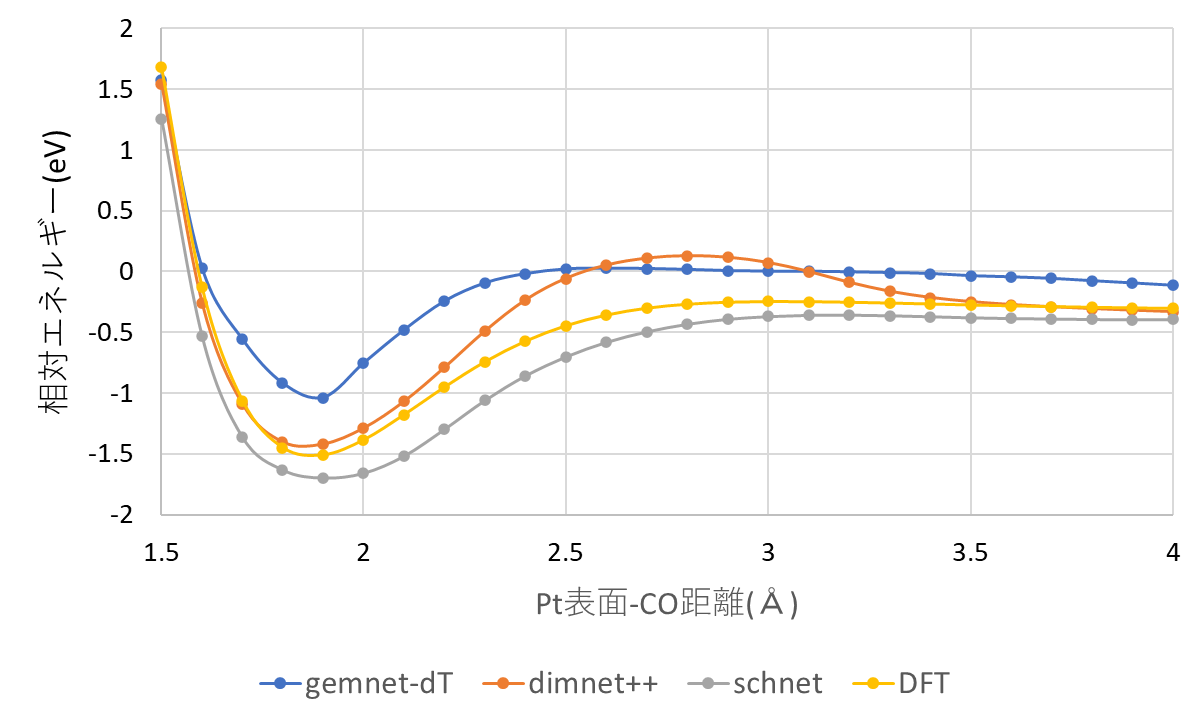
図2に吸着ポテンシャルカーブ計算結果を、表2に吸着エネルギーの計算結果を示します。使用するGNN力場の手法により計算結果に差異は見られるものの、0.5eV以下の精度で吸着エネルギーを予測できていることが分かります。また、1点当たりのエネルギー計算に要した時間を比較すると、GNN力場は第一原理計算と比べて200倍以上高速に計算できることが確認できました。
| 手法 | 吸着エネルギー(eV) | 1点当たりの計算時間(sec)※ |
| Gemnet-dT | -1.037 | 15 |
| Dimnet++ | -1.420 | 15 |
| Schnet | -1.696 | 15 |
| DFT | -1.510 | 4000 |
まとめ
- 汎用型GNN力場を用いることで、第一原理計算と同程度の計算精度で、低コストで高速に触媒材料の計算が可能となります
- 膨大な学習データを用いて構築された力場であるため、広範囲な触媒材料のシミュレーションが可能となります
[1] Chanussot Lowik et.al. "Open Catalyst 2020 (OC20) Dataset and Community Challenges", ACS Catalysis (2020)
[2] Customized LAMMPS(7Aug2019) for Neural Network Potential, by AdvanceSoft Corp. http://www.advancesoft.jp.