本事例では全固体電池に使用される固体電解質中のLiイオンの拡散を、ニューラルネットワークMDを用いて解析した事例を紹介します。第一原理計算を用いて固体中のイオン拡散を解析する場合、通常は第一原理分子動力学法(第一原理MD)を用いてイオンの運動を直接追跡する方法が用いられます[1]。しかし、第一原理MDを用いてイオンの拡散経路を完全に再現するためには、長時間のMD計算の結果を用いてイオン分布の統計的に評価しなければならないため、非常に計算コストがかかります。そこで、本事例では第一原理計算と同程度の計算精度で高速にMD計算を実行できるニューラルネットワークMDを用いて固体電解質中のLiイオンの拡散分布を評価しました。
1.学習データ
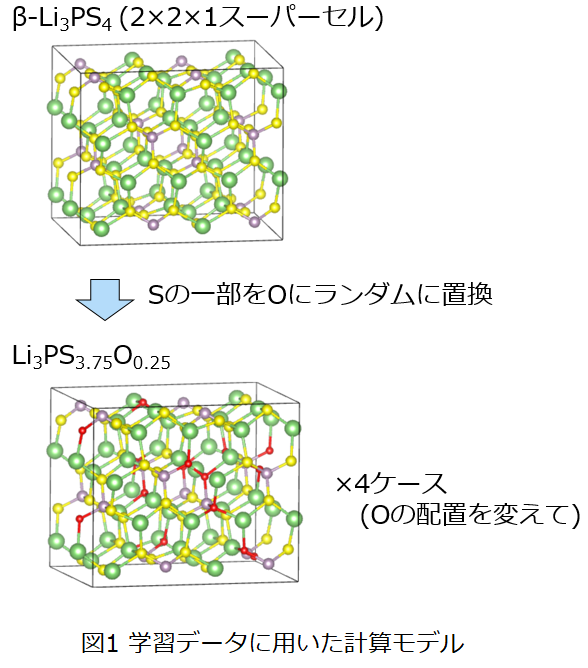
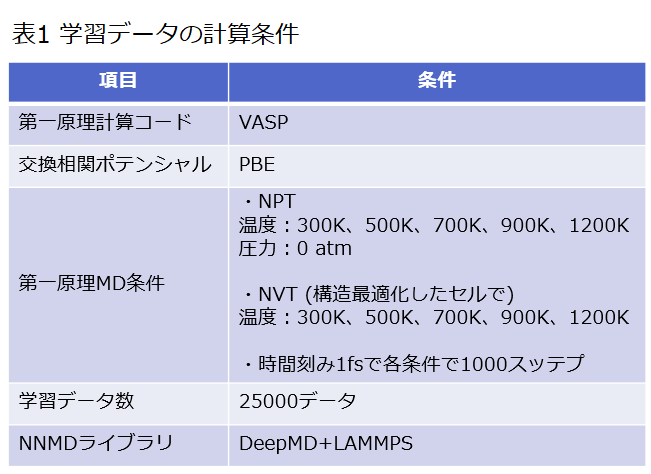
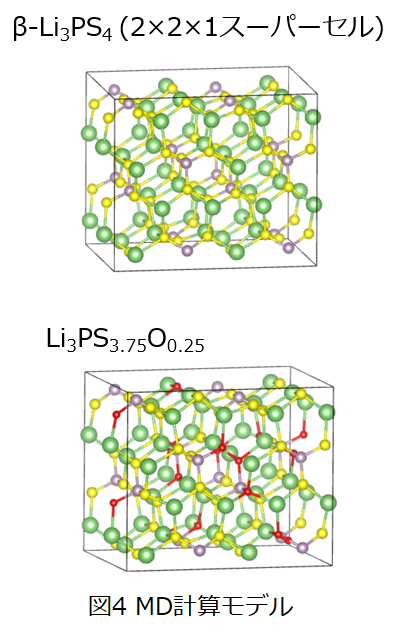
ここでは固体電解質としてLi3PS4、およびLi3PS4に対して一部酸素をドープしたLi3PS3.75O0.25に対してLiイオンの拡散を検討しました。学習データに用いた計算モデルおよび計算条件を図1および表2に示します。学習データは第一原理計算コードVASPを用いて第一原理MDより計算したデータを用いて作成しました。。また、ニューラルネットワークMDライブラリにはDeepMD[2]を用いました。
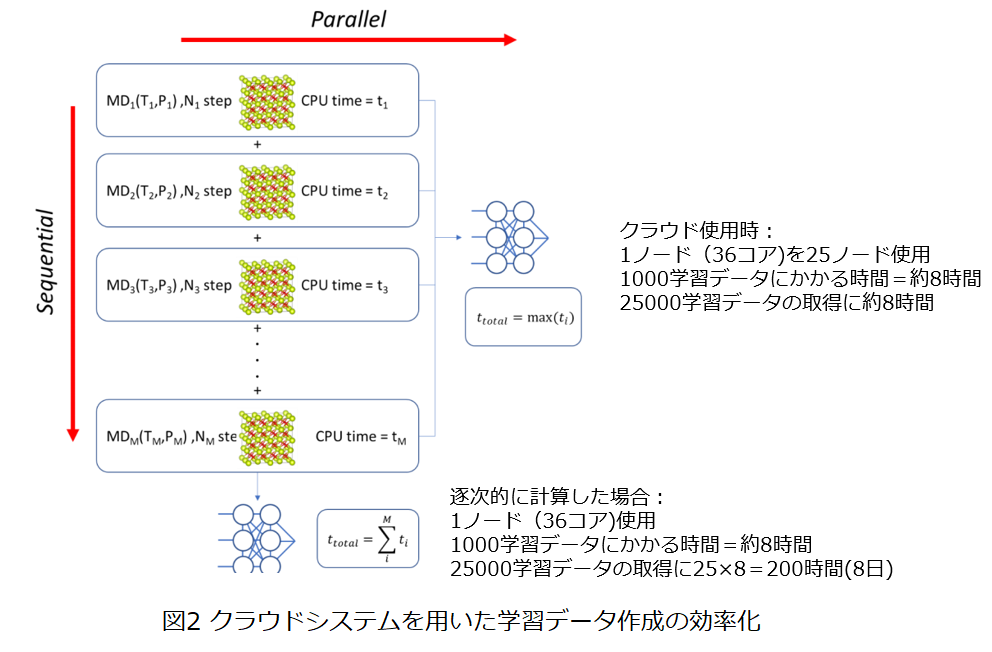
学習データの作成のために、広範囲の温度・圧力条件を課した第一原理分子動力学計算を多数回実行する必要があるため、、 通常のクラスター計算機環境で逐次的に計算を実行すると膨大な計算時間が必要となりますが、 クラウドシステムを使用することで、一度に全ての第一原理計算度同時に実行して短時間でポテンシャル学習を行うことができます(図2)。
2.ポテンシャル学習結果
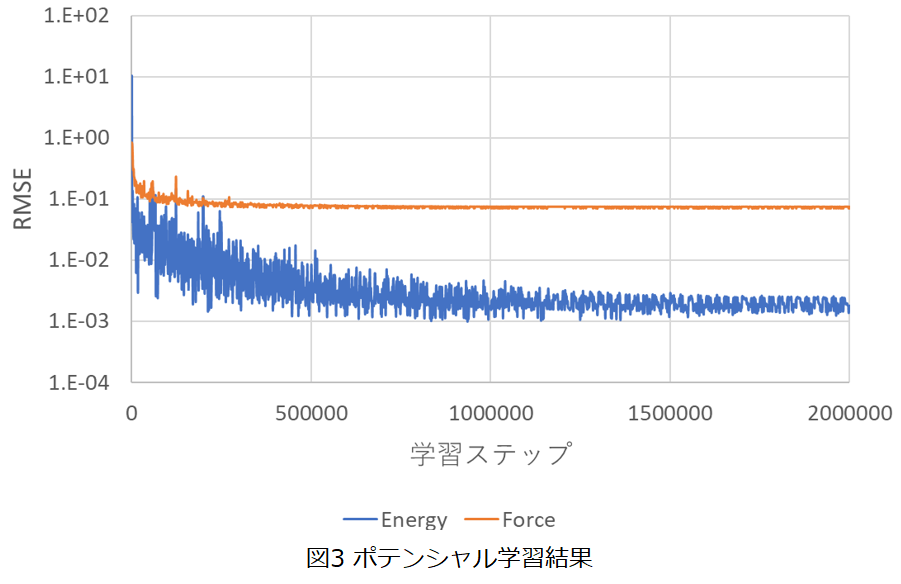
図3にDeepMDを用いたポテンシャル学習結果を示します。第一原理計算結果に対して、エネルギーは約0.002eV、原子に加わる力は約0.07eV/Åの誤差で学習できています。
また、第一原理MDとニューラルネットワークMDの計算時間を比較すると、MD1000ステップに要する時間は36コア使用時で、第一原理MDでは約12000秒であるのに対してニューラルネットワークMDでは90sとなり、ニューラルネットワークMDを使用することで100倍以上高速に計算できていることが確認できました。
3.MD計算条件
作成したニューラルネットワークポテンシャルデータを用いてLi3PS4、およびLi3PS3.75O0.25におけるLiイオンの拡散係数を評価しました。図4に使用した計算モデルを、表2に計算条件を示します。全ての分子動力学計算にはLAMMPSを用いています。 また、拡散係数はエネルギー一定分子動力学計算(NVE)で得られたLiイオンの平均二乗変位を用いて以下の式により評価しました。
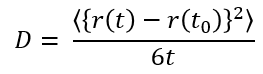

4. 計算結果
左記にLi3PS4、およびLi3PS3.75O0.25のMD計算結果のアニメーションを示します。 高温(600K)となり高イオン伝導状態となると、Liイオンは格子間をジャンプする規則的な伝導経路をたどらず、不規則に分布している様子が確認でき、Li-PS型電解質の実験結果を再現する結果となりました。
また、図5にLiイオンの平均二乗変位の計算結果を示します。Li3PS4、およびLi3PS3.75O0.25の結果を比較すると、酸素ドープによりLiイオンの平均二乗変位が増加することが分かります。 また、シミュレーション時間が50ps以上にならないと低温状態における平均二乗変位の増加傾向が確認できないため、第一原理計算では低温状態のイオン伝導性を評価することは困難となりますが、ニューラルネットワークMDではns程度の長時間シミュレーションも実行可能なため、低温状態のLiイオン伝導性を評価することが可能となります。
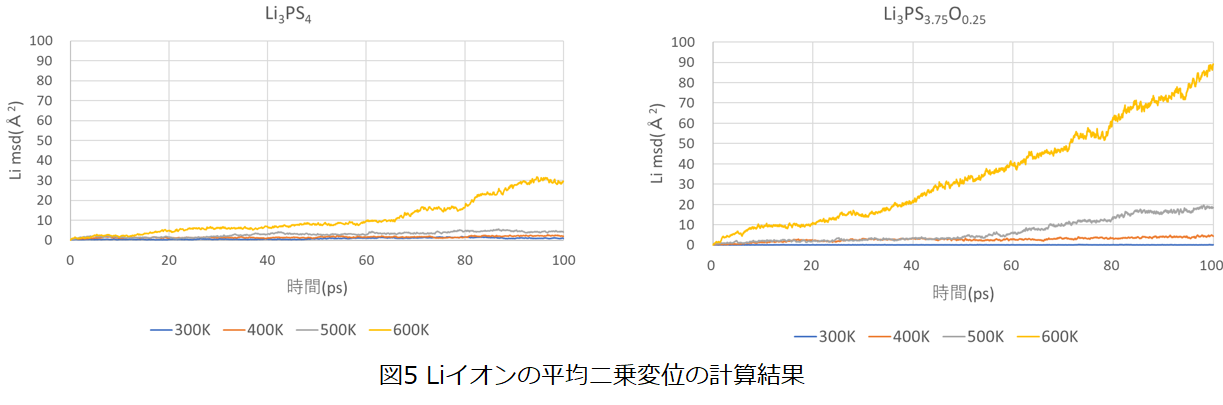
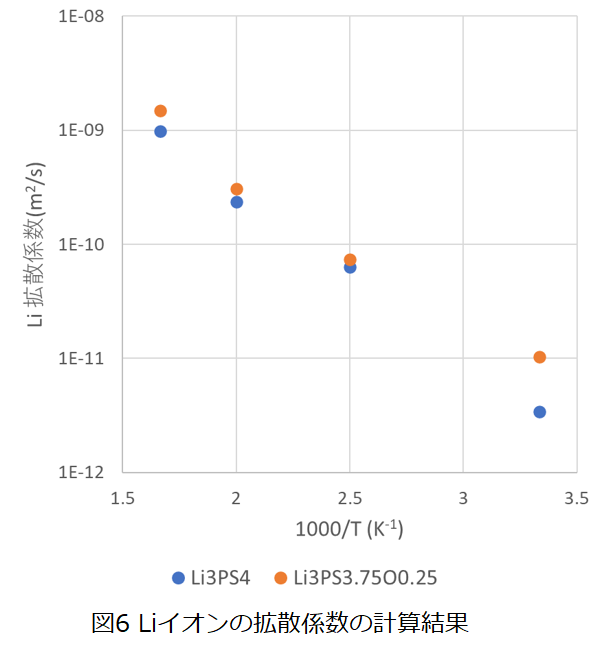
図5で得られた平均二乗変位の計算結果よりLiイオンの拡散係数を評価した結果を図6に示します。 Li3PS4、およびLi3PS3.75O0.25の結果を比較すると酸素ドープによりLiイオンの拡散係数が増加し、イオン伝導性が向上する可能性があることが確認できました。
5.今後の展開
本手法を応用することにより、以下の展開が期待できます。
① MIと連携したイオン伝導性とドーパント種、ドーパント濃度の最適化
② 電極/電解質界面を考慮したニューラルネットワークMDによる界面抵抗の評価
6.参考文献
[1]R. Xiao, H. Li and L. Chen, “High-throughput design and optimization of fast lithium ion conductors by the combination of bond-valence method and density functional theory”, Scientfic Reports, 5, 1 (2015)
[2]Han Wang, Linfeng Zhang, Jiequn Han, and Weinan E. “DeePMD-kit: A deep learning package for many-body potential energy representation and molecular dynamics.” Computer Physics Communications 228 (2018): 178-184.