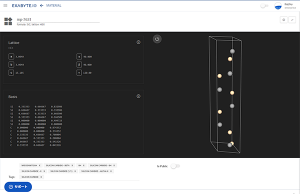
NEB計算により、白金Ptの111表面上を移動する酸素O原子の活性化エネルギーを求めることができます。
Pt表面にO原子を設置したモデルは、Exabyte.ioのMulti-Material 3D Editorを使用することで作成できます。hollow-HCPサイトとhollow-FCCサイトにO原子を配置したモデルを作成し、各々構造最適化を行ないます。

図1 hollow-HCPサイト(a) とhollow-FCCサイト(b) に酸素原子を配置したモデル
次にMaterials DesignerのInterpolated Setを使って、NEB計算用のモデルを作成します。ここではinitialをhollow-HCPサイト、finalをhollow-FCCサイトにして、中間のイメージモデルを3つ作成しています。作成したモデルを保存し、workflowのNudged Elastic Band(NEB)を用いてNEB計算を行います。
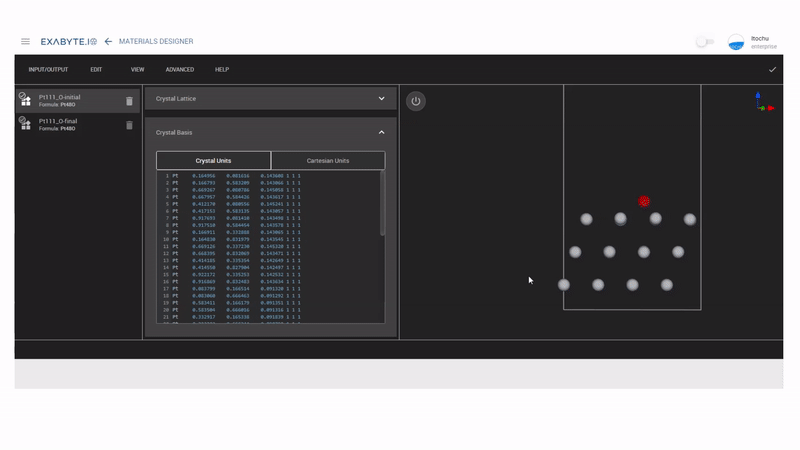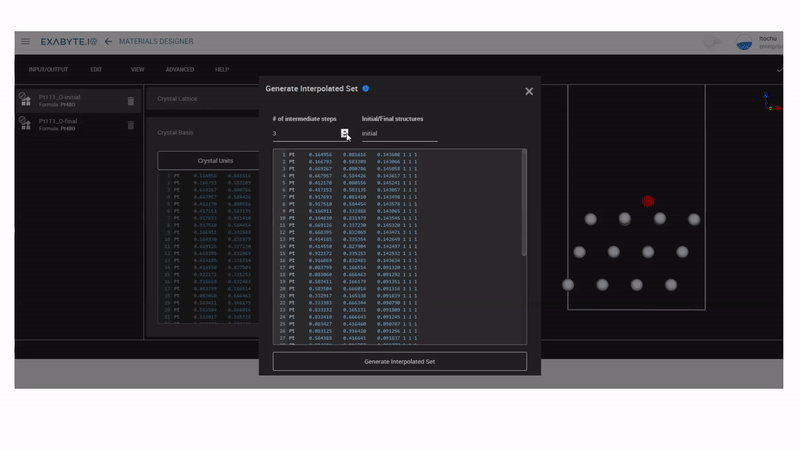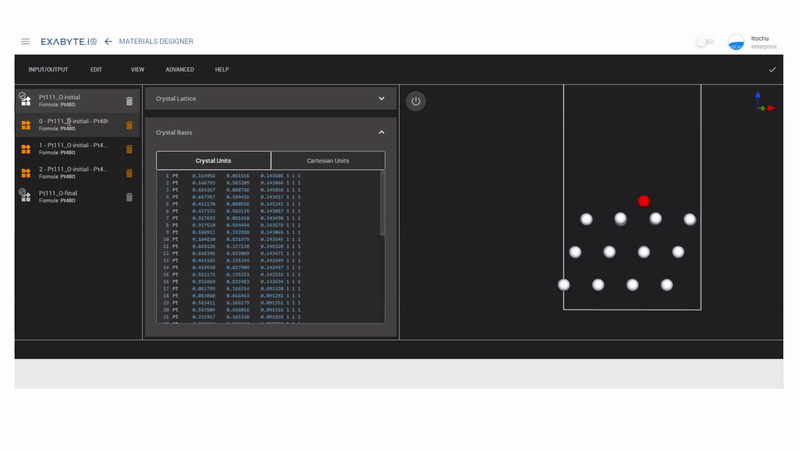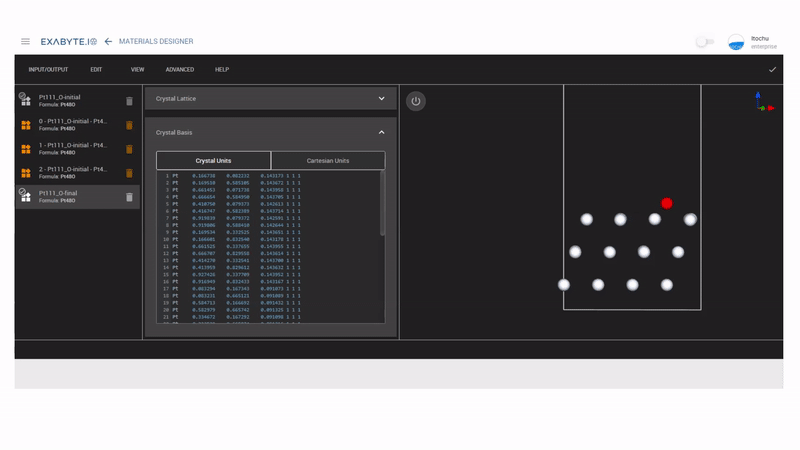
図2 NEBモデル作成の様子
表1 計算条件
| 項目 | 詳細 |
|---|---|
| 計算ソフト | Quantum ESPRESSO |
| 擬ポテンシャル | Pt_pbe_gbrv_1.4.upf O_pbe_gbrv_1.2.upf |
| カットオフ 波動関数 | 40 Ry |
| カットオフ 電子密度 | 200 Ry |
| k点 | 3 × 3 × 1 |
| 収束閾値 | 10-6 |
| mixingパラメータ | 0.3 |
| 原子数 | 49原子 Pt:48 O:1 |
| CPU | Intel® Xeon® CPU CPU E5-2667 v3 @ 3.20GHz |
| コア数 | 15 core |
NEB計算により、hollow-HCPサイトからhollow-FCCサイトにO原子が移動する際のエネルギー曲線が求まります。
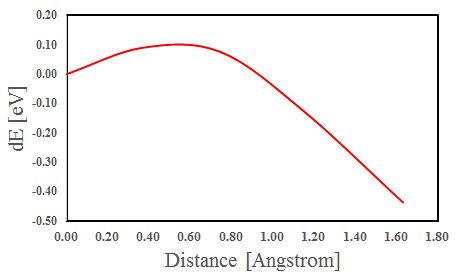
図3 hollow-HCPサイトからhollow-FCCサイトにO原子が移動する際のエネルギー曲線
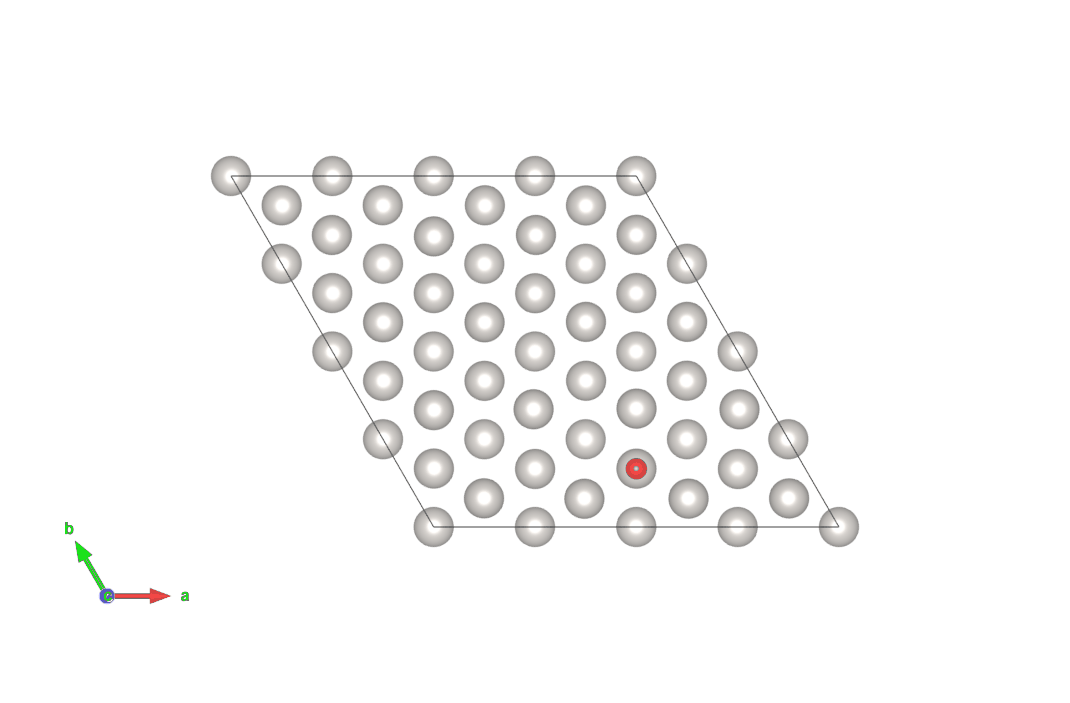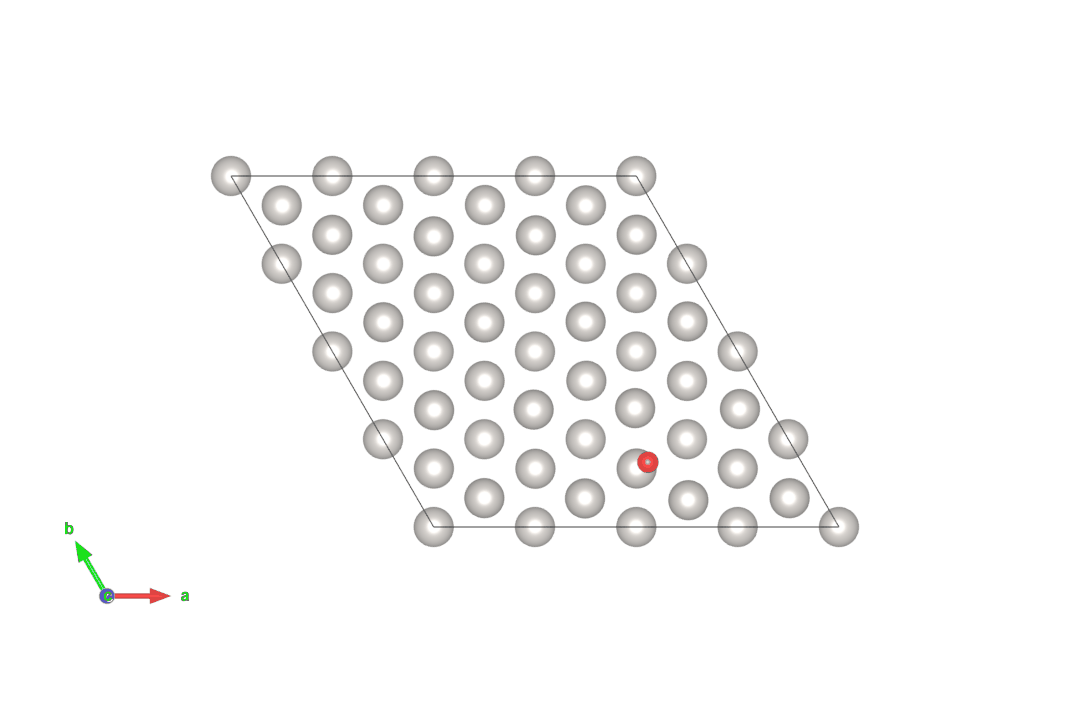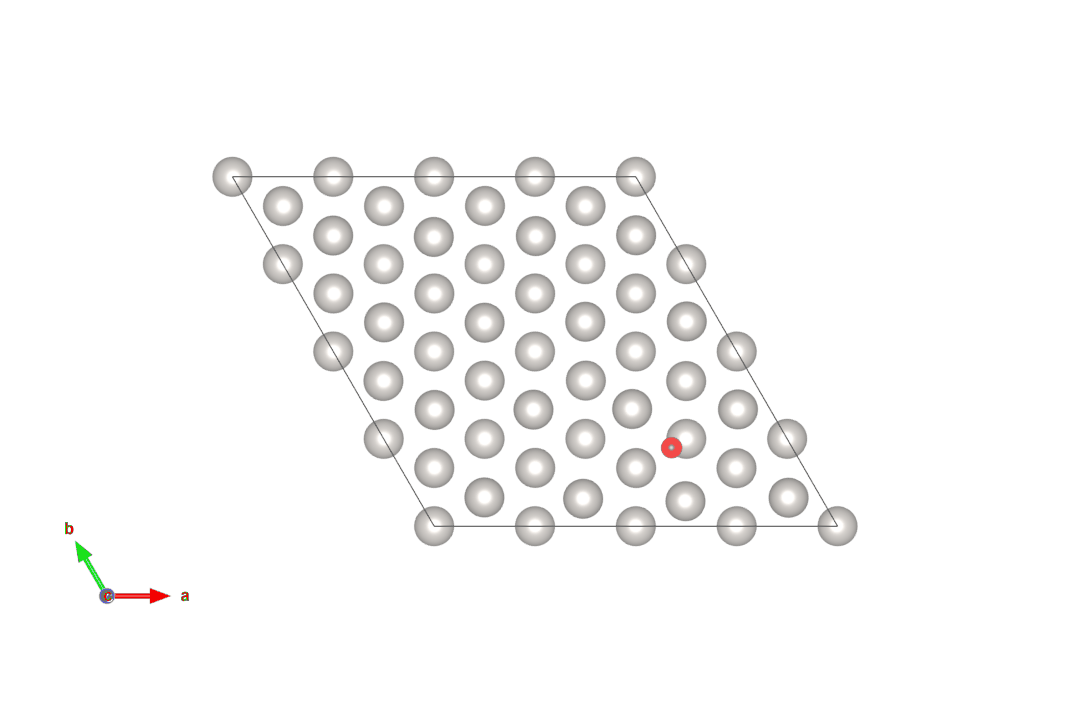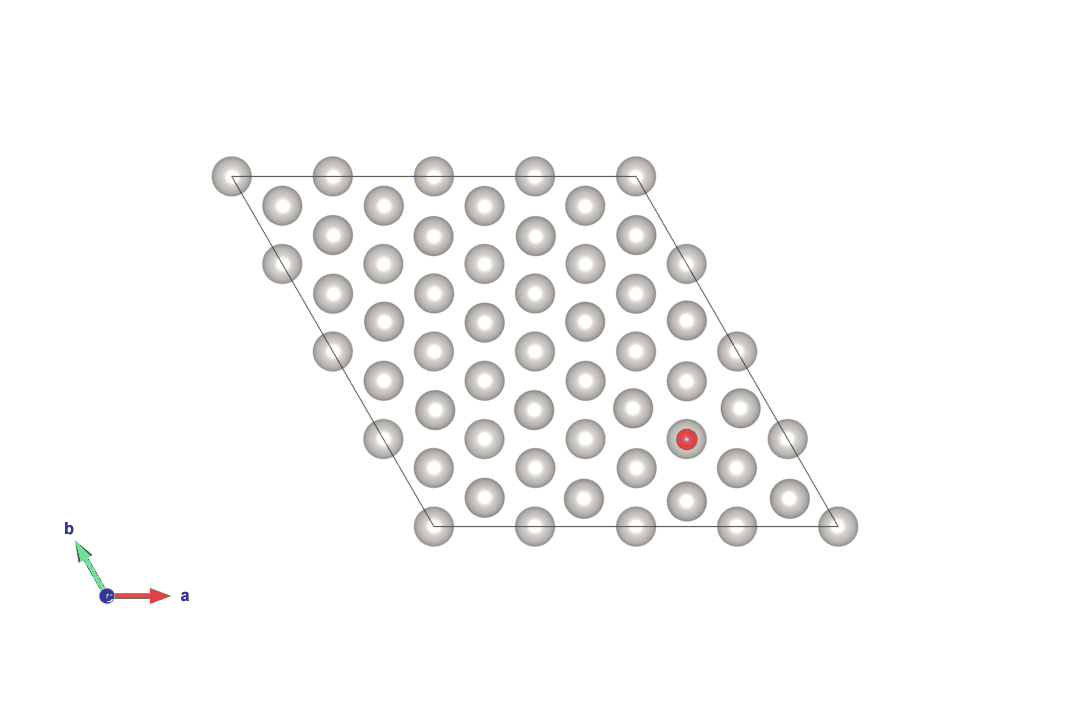
図4 hollow-HCPサイトからhollow-FCCサイトにO原子が移動する計算結果(上)
画像クリックでアニメーションとなります。

図5 hollow-HCPサイトからhollow-FCCサイトにO原子が移動する計算結果(側面)
画像クリックでアニメーションとなります。
計算から求められたエネルギー障壁は0.53 eVでした。先行研究で計算された値は0.5 eVであり、今回の計算結果を近い値になっています[1]。実験値は0.43 eVで、計算値は実験値と近い値になっています[2]。
参考文献
[1] Pang Q, Zhang Y, Zhang J M, et al. Structural and electronic properties of atomic oxygen adsorption on Pt (111): A density-functional theory study. Appl Surf Sci, 2011, 257: 3047-3054
[2] J. Wintterlin, R. Schuster, and G. Ertl, Existence of a “Hot” Atom Mechanism for the Dissociation of O2 on Pt (111), Phys. Rev. Lett. 1996, 77, 123 - 126