概要
近年注目されている異材接合技術をはじめとした、様々な産業技術において、樹脂/金属酸化物界面の微視的挙動を理解することは重要です。材料の微視的挙動の解析にはしばしば分子動力学法(MD)が用いられますが、樹脂/金属酸化物の相互作用を精度良く表す力場がないためMDの適用が制限されています。そこで、本記事ではDFT計算で得られたエネルギーへのフィッティグにより、樹脂/金属酸化物間のMD力場を新たに開発した事例を紹介します。本事例ではα-Al2O3/PA6、α-Fe2O3/PA6の力場を開発し、引張強度、界面エネルギーを評価しました。
力場パラメータの最適化
DFT計算で得られたエネルギーへのフィッティグにより、樹脂/金属酸化物間のMD力場を最適化しました。図1のように金属酸化物上にPA6モノマーを三種の方向で置き、酸化物と樹脂の距離を変えたときのDFT計算によるエネルギー変化を、MD力場で再現できるよう力場パラメータを決定しました。
DFT計算にはVASPを使用しました。VASPを用いた計算では、PA6の初期位置を構造最適化計算により決定し、酸化物-樹脂間の距離を変えたモデルのエネルギーはSCF計算で計算しました。なお、DFT計算におけるvan der waals相互作用の補正にはGrimmeのDFT-D2法を使用しました。
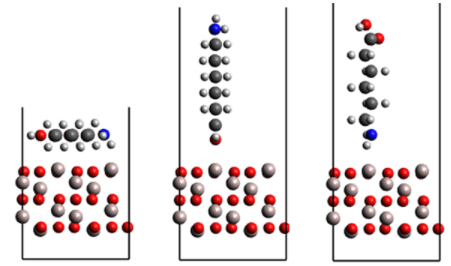
図1 力場パラメータ最適化のための計算モデル
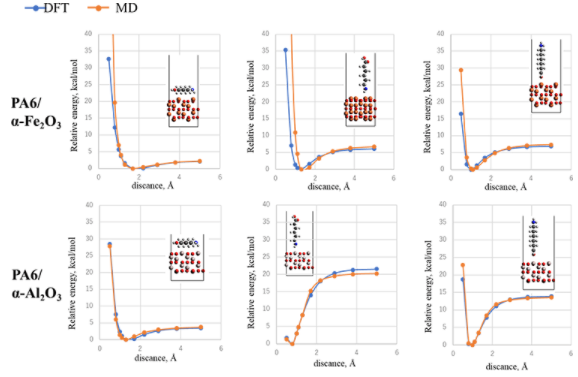
図2 フィッティング結果
表1 使用したMD力場
| MD力場 | |
|---|---|
| PA6 | OPLS-AA (PolyParGenにより作成[1]) |
| Fe2O3 | Coulomb-Buckingham potential(文献値[2]) |
| Al2O3 | Coulomb-Buckingham potential(文献値[3]) |
| PA6/Fe2O3, PA6/Al2O3 | Coulomb-LJ potential + Coulomb-Morse potential (本事例にて開発。官能基-酸化物間でMorse potentialを使用) |
MD計算
開発したパラメータを使用し、MD法による界面剥離シミュレーションを行いました。樹脂を酸化物で挟んで十分緩和させた後、酸化物に一定の速度をあたえるシミュレーションを行いました。


図3 剥離シミュレーション (左) A6/Al2O3 、(右) PA6/Fe2O3
シミュレーションに用いたPA6の重合度及び分子数を示します。
表2 PA6の重合度及び分子数
| 重合度n | 10 |
|---|---|
| 分子数 | 556 |
| 原子数 | 107308 |
シミュレーションに用いた金属酸化物モデルを示します。なおα-Al2O3、α-Fe2O3はいずれもコランダム構造です。

図4 金属酸化物モデル
シミュレーションに用いたソフト及び計算条件を示します。
表3 計算条件
| 計算ソフト | Lammps |
|---|---|
| 力場 | 表1を参照 |
| タイムステップ | 1 fs |
| アンサンブル | NVT |
| 温度 | 300 K |
| 温度制御 | Nose-Hoover |
| 歪み速度 | 6.67×109 /sec |
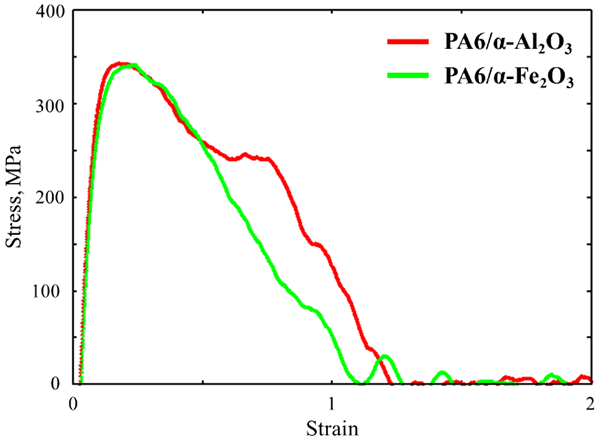
図5 応力ひずみ曲線
表4 引張計算前の樹脂/金属界面エネルギー
| 界面エネルギー, mJ/m2 | |
|---|---|
| PA6/α-Al2O3 | -3399.20 |
| PA6/α-Fe2O3 | -2172.86 |
以上のように樹脂とスラブの組み合わせ方に応じて界面の力場パラメータ開発することで、界面の組み合わせの違いによる微視的挙動の違いを、MD解析することが可能となります。
参考文献
[1] M. Yabe, K. Mori, K. UeDa, M. TaKeDa, J. Comput. Chem. Jpn. Int. Ed. 2019, 2018-0034
[2] TD Ta, University of Woollongong Thesis Collection 1954-2016
[3] Z. Hu, J. Shi and C. H. Turner, Molecular Simulation, 2009, 270-279