汎用型グラフニューラルネットワーク力場(GNN力場)を用いて触媒材料の分子動力学シミュレーションを行いました。用いたGNN力場は、Facebook社とカーネギーメロン大学が主催するOpen Catalyst Projectで開発されたもので、触媒材料をターゲットとして、約1億3千万個の第一原理計算の計算結果を学習データとして構築されており、広範囲な触媒材料の分子動力学計算が可能となっています。本事例では、Ni系触媒を用いたCO2メタネーション反応の解析を行いました。
計算モデル・計算条件
CO2メタネーション反応式はCO2 + 4H2 → CH4 + 2H2Oですが、ここでは以下の反応過程に示すように、水素分子は触媒表面に解離吸着して水素原子となり、CO2分子へ水素付加することで進行するものと仮定しました。
| CO2 + H → HCO2 | (1) |
| HCO2 + H → HCOOH | (2) |
| HCOOH + H → H2COOH | (3) |
| H2COOH + H → H2CO + H2O | (4) |
| H2CO + H → H2COH | (5) |
| H2COH + H → H2COH2 | (6) |
| H2COH2 + H → H3COH2 | (7) |
| H3COH2 + H → CH4 + H2O | (8) |
触媒モデルは、Ni(111)面とし、さらにNi原子の一部をMgおよびFeに置換した場合についても検討しました。反応開始時の計算モデルを図1に示します。化学反応の計算にはNudged elastic band(NEB)法を用いました。NEB法は反応の開始状態と終状態の構造から遷移状態を探索する方法です。また、使用したGNNはDimnet++としました。

計算結果
図2にNEB計算より得られたメタネーション反応のアニメーションを示します。水素付加が進行するに従い、水分子が脱離して最終的にメタン分子が生成していることが分かります。第一原理計算を用いてこのようなNEB計算を実行すると非常に膨大な計算時間を要しますが、ここでは1反応過程のNEB計算に要する時間は数分程度で、非常に高速に計算が終了しています。
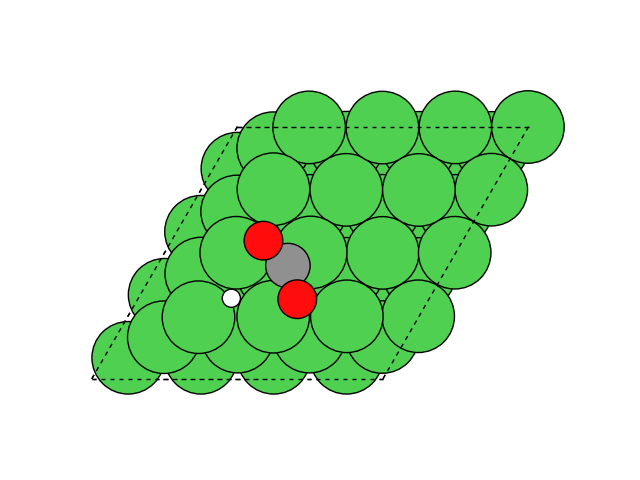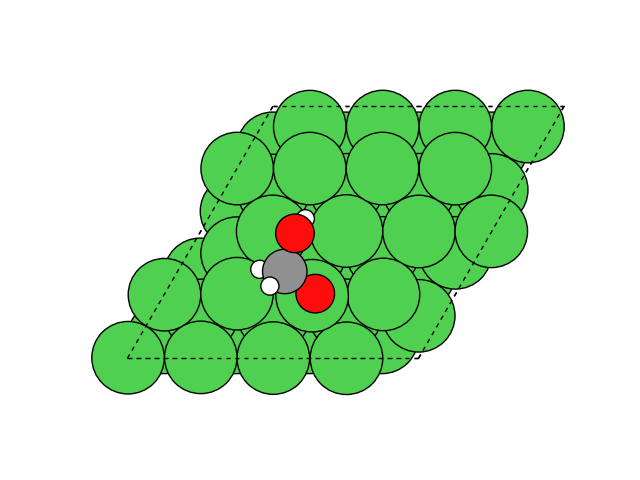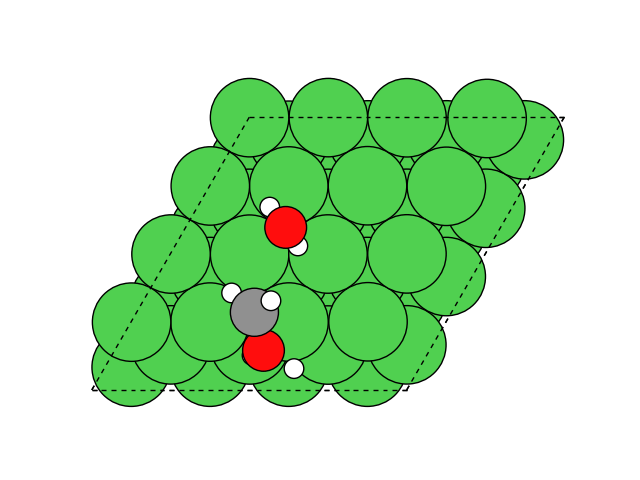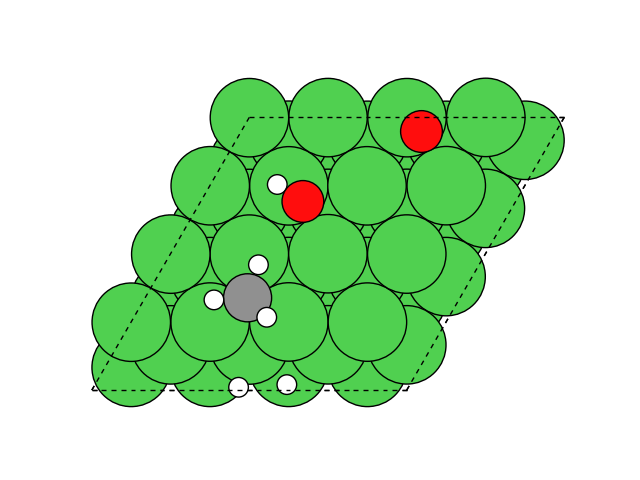
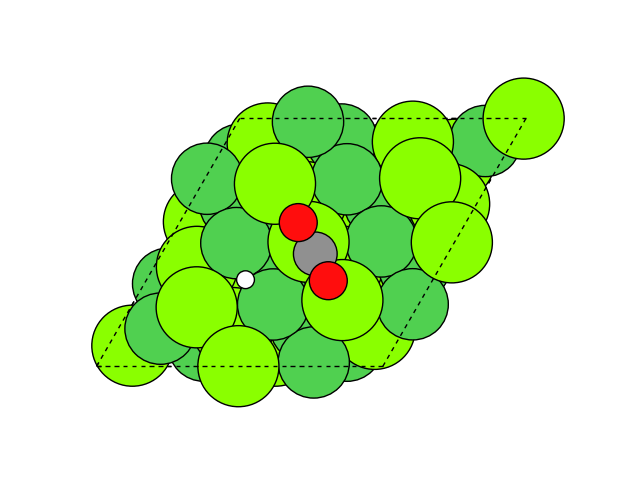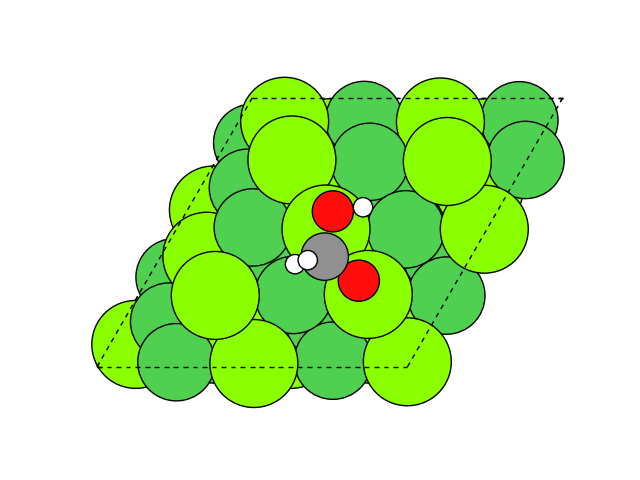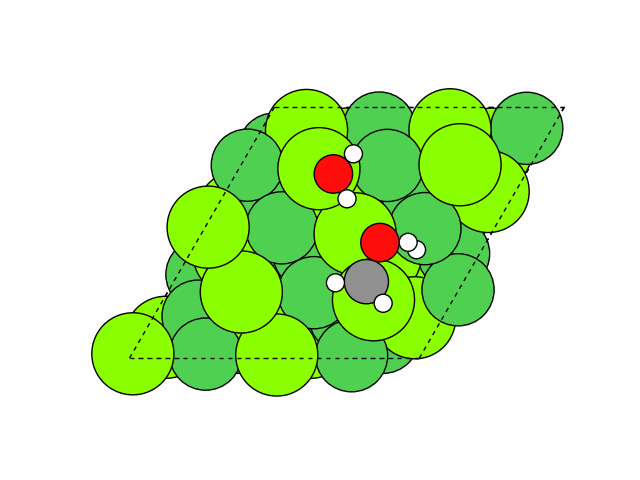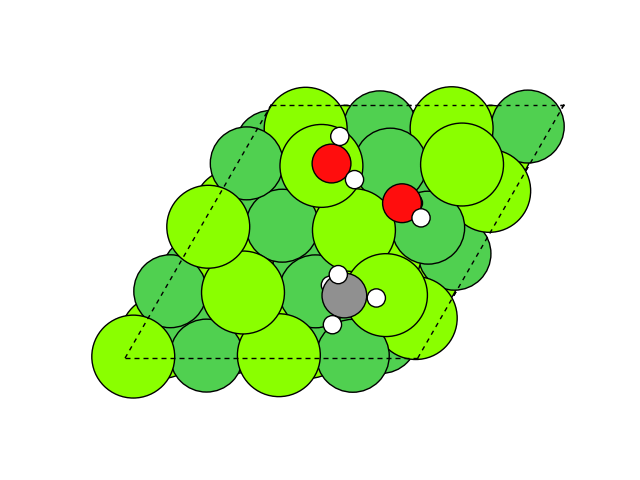
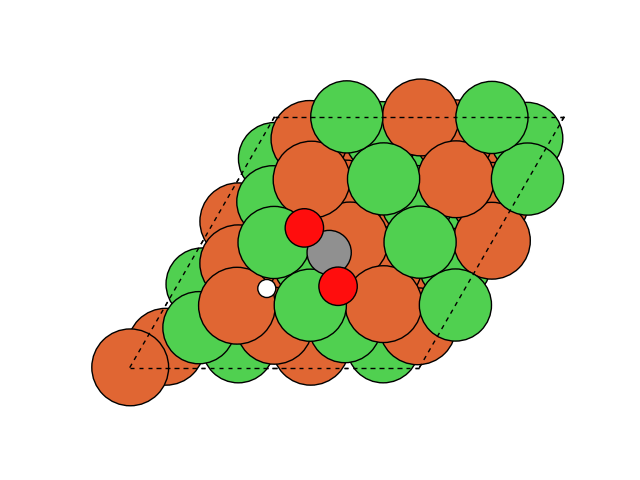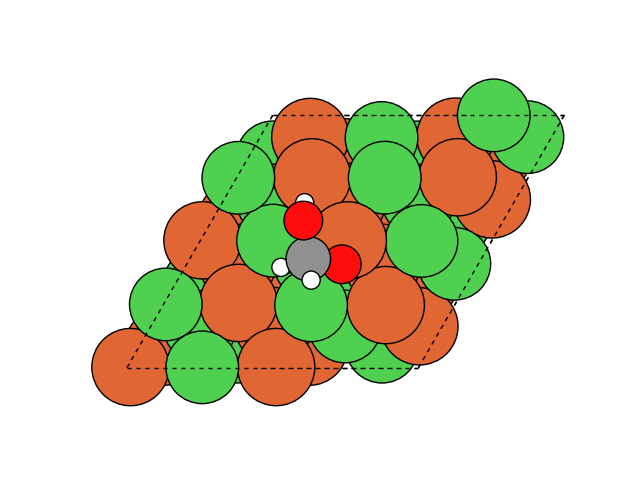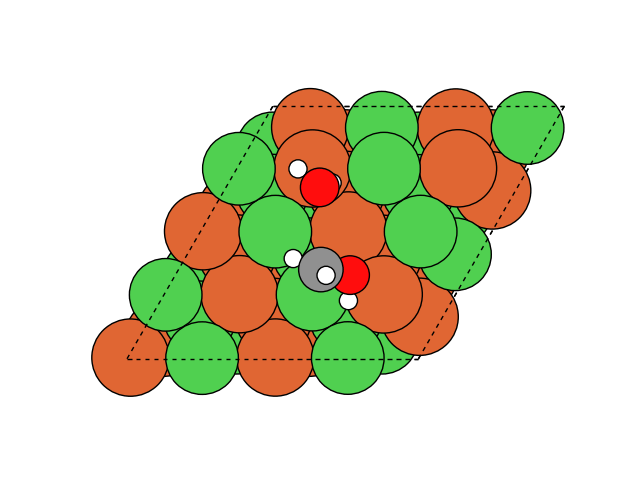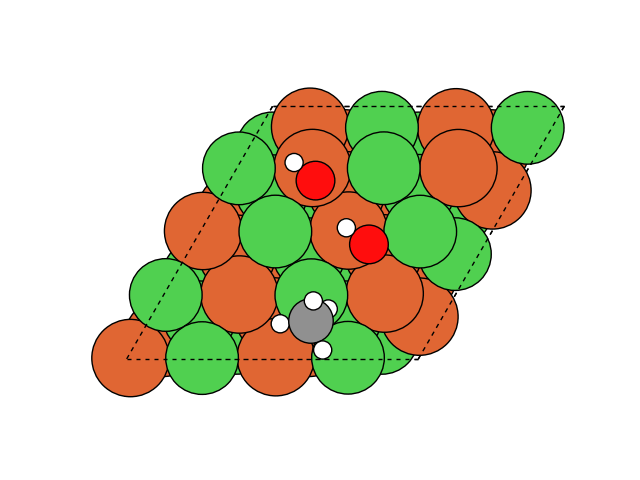
図2 メタネーション反応過程のシミュレーション結果
図3に反応のプロファイルを示します。反応の律速過程は水分子が脱離する過程であることが分かりました。律速過程の活性化エネルギーを表2に示します。NiをMgに置換するとわずかに活性化エネルギーが減少することが確認できました。
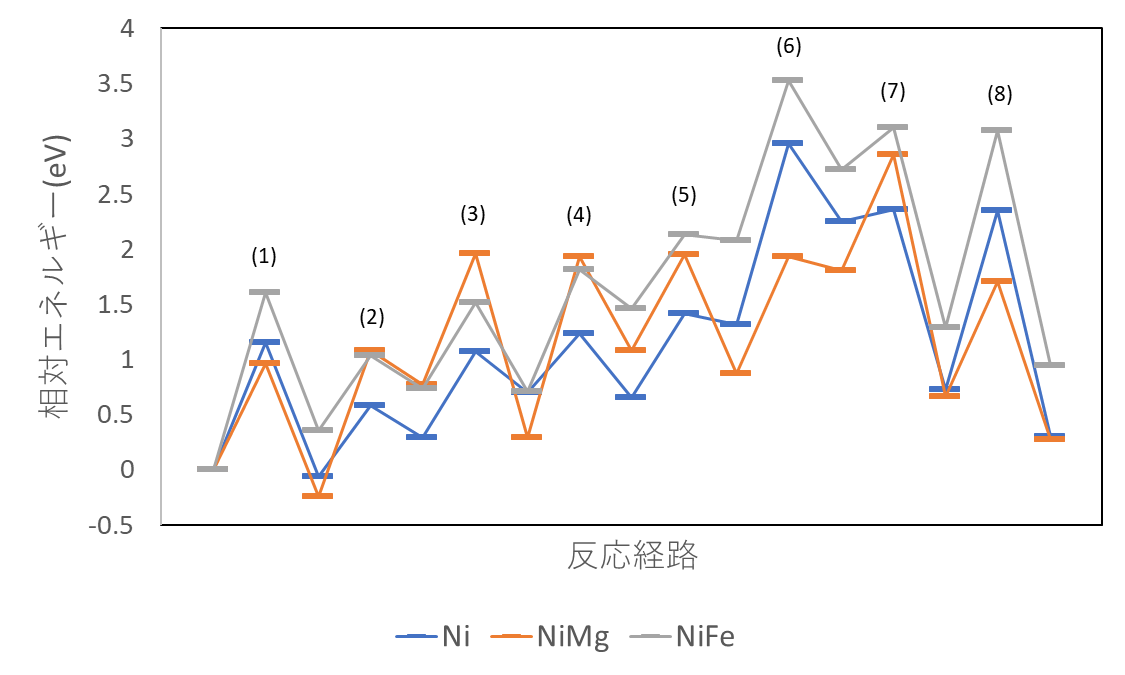
| モデル | 活性化エネルギー | 律速過程 |
| Ni | 1.65 eV | (8) |
| NiMg | 1.63 eV | (4) |
| NiFe | 1.79 eV | (8) |
まとめ
汎用型GNN力場を用いて触媒反応の解析が可能であることが確認できました。
第一原理計算では膨大な計算時間を要する化学反応探索が、汎用型GNN力場を用いると数分程度で終了し、高速に化学反応探索が可能となります。